Revisión
Enfermedades por priones: de la clínica a la biología molecular
Prion diseases: from molecular biology to clinical practice
Carlos Andrés Villegas Lanau
Carlos Andrés Villegas Lanau. Médico. Magíster en Ciencias Básicas Biomédicas. Doctorado en Ciencias, Universidad de Antioquia. Profesor del Instituto de Investigaciones Médicas, Facultad de Medicina de la Universidad de Antioquia. Investigador del Grupo de Neurociencias de Antioquia.
Correo electrónico: andres.villegas@neurociencias.udea.edu.co
Recibido: 1/02/10. Revisado: 7/02/10. Aceptado: 10/05/10.
RESUMEN
Las enfermedades ocasionadas por priones son también conocidas como encefalitis espongiformes transmisibles o demencias de tipo infeccioso. En humanos las presentaciones clínicas más reconocidas son la enfermedad de Creutzfeldt-Jakob, el síndrome de Gerstmann-Sträussler-Scheinker y el insomnio fatal familiar. Son consideradas patologías poco comunes, pero los descubrimientos en biología molecular de los últimos años muestran que los mecanismos patológicos que llevan a su desarrollo pueden ser comunes a varias enfermedades neurodegenerativas, lo cual puede ampliar el espectro patológico, convirtiéndolas en alteraciones no tan infrecuentes en neurología.
Esta revisión pretende dar herramientas al clínico para reconocer estas enfermedades discutiendo las presentaciones clínicas en seres humanos con sus variantes: esporádicas, infecciosas y familiares, comentando además el uso de laboratorios, criterios diagnósticos y aproximación terapéutica.
PALABRAS CLAVES. Prión, biología molecular, encefalitis.
SUMMARY
Illnesses caused by prions are also known as Transmissible Spongiform Encephalopathies or infectious dementias. The most recognized clinic presentations in humans are the Creutzfeldt-Jakob disease, the Gerstmann-Sträussler-Scheinker syndrome and the Fatal Familial Insomnia. They are considered uncommon pathologies, but discoveries in molecular biology in recent years, reveal that pathological mechanisms that allow its development could be common to several neurodegenerative diseases in which the pathologic spectrum can be amplified, becoming not infrequent neurological diseases. This review gives tools to the clinical attendant in order to recognize these disorders, discussing the clinical presentations: sporadic, infectious and familiar, diagnostic work up, diagnostic criteria and therapeutic approach.
KEY WORDS. Prions, molecular biology, encephalitis.
INTRODUCCIÓN
Hace más de 200 años fue descrita en Inglaterra una enfermedad que afecta a ovejas y cabras, y que por su síntoma de rascado intenso, el cual les provoca laceraciones y pérdida de la lana, fue llamada scrapie (rascar, en inglés). Esta enfermedad se acompaña de ataxia progresiva y temblor, y es fatal en todos los casos (1). Cuando se identificó el scrapie como una enfermedad infecciosa se trató de identificar el agente etiológico causal, encontrando que se trataba de uno filtrable, con un tiempo muy prolongado de incubación y capaz de soportar tratamientos con calor, formaldehído y radiación que normalmente destruyen a los agentes infecciosos conocidos, de manera que fue clasificado inicialmente como virus lento o agente no convencional (2, 3). El hallazgo de que el agente del scrapie carece de ácidos nucleicos llevó a John Stanley Griffith a proponer que la enfermedad es producida por una proteína que se comporta como un agente infeccioso (4). Carleton Gajdusek fue el primero en identificar una enfermedad en humanos cuyo agente causal tiene las mismas características que las encontradas en el scrapie; fue identificada como kuru, una patología que afecta a la tribu Fore, la cual habita las tierras altas de Papúa, en Nueva Guinea, e identificó la causa de la transmisión de la enfermedad por un rito caníbal desarrollado en esta comunidad (5). Estos descubrimientos condujeron a identificar otras patologías, diferentes al kuru, en humanos, causadas por agentes con las mismas características del scrapie, como la enfermedad de Creutzfeldt-Jakob, y posteriormente, el síndrome de Gerstmann-Sträussler-Scheinker (1).
Fue Stanley Prusiner quien desarrolló la hipótesis priónica, postulando que una proteína llamada priónica (PrP) puede adoptar dos configuraciones distintas, y una de estas conformaciones puede llevar al cambio de conformación de la otra proteína, lo cual produce un comportamiento de la proteína que provoca el cambio conformacional, similar al de un agente infeccioso (6). Prusiner también acuñó el término prión (acróstico de proteinaceous infectious agent), descubrió el gen codificador para la proteína priónica que normalmente se encuentra en las células (PrPC) y desarrolló modelos transgénicos que ayudaron a demostrar su hipótesis (7).
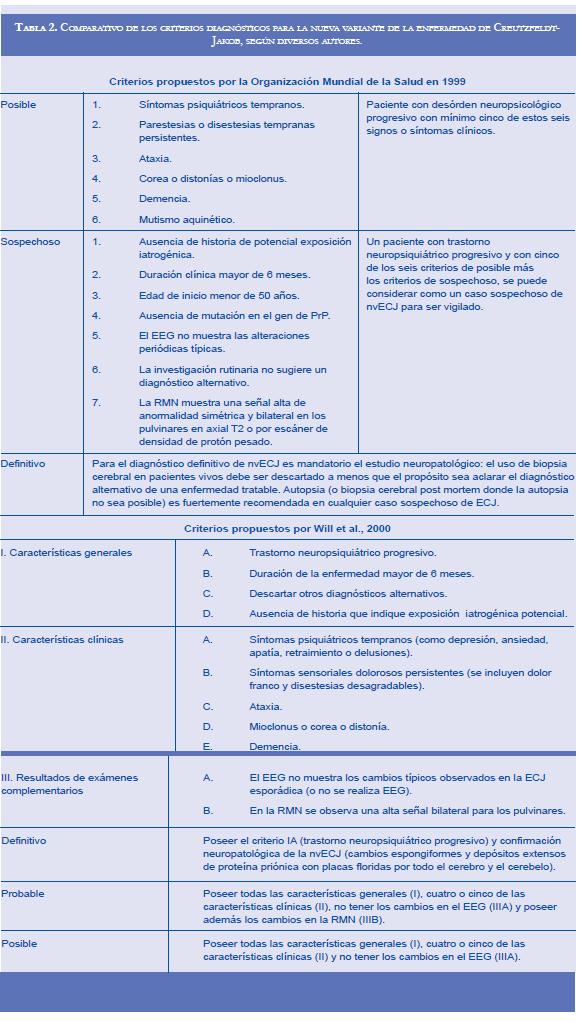
Aunque la enfermedad de Creutzfeldt-Jakob (ECJ) se conocía desde la década de los años veinte del siglo pasado, cuando el neurólogo Hans Gerhard Creutzfeldt describió el primer caso y luego Alfons Maria Jacob reportó cinco casos más, esta enfermedad descrita por ellos ha sido llamada de la forma clásica, para diferenciarla de la nueva variante (nvECJ) (8). La nueva variante de ECJ tuvo su origen en Inglaterra en 1995, donde fueron descritos los primeros dos casos. En la actualidad existe una fuerte evidencia sobre que la nvECJ fue producida por el consumo de carne vacuna contaminada con encefalitis espongiforme bovina (EEB), y a su vez la EEB tuvo su origen al cambiar el procesamiento en la producción de harinas de engorde para el ganado en las décadas de los setenta y los ochenta, llevando a la aparición de la enfermedad (EEB), descrita en 1988, y a la respectiva epidemia de EEB (vulgarmente llamada enfermedad de las vacas locas), que tuvo su pico máximo en 1992, con 170.000 reses afectadas (9).
En general las enfermedades causadas por priones poseen características comunes, como son: pérdida progresiva de neuronas, reducida respuesta inflamatoria, vacuolización del neuropilo (provocando la llamada encefalopatía espongiforme), depósitos de proteína priónica con conformación anómala y la capacidad de transmitir la enfermedad (por lo que también son llamadas encefalopatías espongiformes transmisibles). Además se ha identificado a las enfermedades por priones en humanos como demencia infecciosa, debido al cuadro de demencia rápidamente progresiva que las caracteriza (10). En esencia, se identifica a un prión como una proteína que por un cambio en su conformación (transformación de hélices alfa en pliegues beta) cambia sus propiedades físico-químicas, haciéndose más resistente a agentes que normalmente la degradarían y formando agregados proteicos; adicionalmente, adquiere la capacidad de provocar en proteínas iguales a ella el mismo cambio conformacional, convirtiéndolas en un agente transmisible, esto es, puede pasar de un individuo a otro. En mamíferos sólo se conoce una proteína con esas características, llamada PrP, pero se especula sobre otras que parecen tener las mismas características (11).
En humanos el gen que produce la proteína priónica se encuentra en el brazo corto del cromosoma 20, en el sitio 20p12.17, ocupando el locus llamado PRNP; este gen codifica para una proteína de 253 aminoácidos. En su forma normal (PrPC) tiene notables diferencias con la conformación patológica (PrPSc), entre las cuales se encuentran: la PrPC no es infecciosa, en su estructura predominan las hélices alfa, es fácilmente soluble en agua (hidrofílica), sensible a proteasas, e interactúa con anticuerpos anti-PrP, mientras que la PrPSc es infecciosa, en su estructura abundan los pliegues beta, es insoluble en agua, posee alta resistencia a proteasas y tiene poca reactividad a los anticuerpos anti-PrP (12). La PrP posee una región en la cual ocho aminoácidos (octapéptido) se repiten cinco veces, esta región es un sitio donde se pueden presentar variaciones en el número de repeticiones de octapéptidos y llevar al desarrollo de formas familiares muy poco comunes de enfermedad en humanos. Adicionalmente, en el codón 129 de la proteína puede haber una sustitución del aminoácido metionina por valina (polimorfismo) y este polimorfismo producir variaciones en la presentación clínica de la enfermedad. Se ha observado que los individuos homocigóticos M/M para este polimorfismo poseen un riesgo más alto para desarrollar las formas esporádicas e infecciosas de la ECJ (13).
Cerca del gen PRNP (16 kilobases escalera abajo) se encuentra un gen homólogo al PRNP que ha sido llamado PRPD (prion protein doblet) y codifica para una proteína denominada doppel o PrPLP (prion protein-like protein), la cual posee 179 aminoácidos. Aún se desconoce la función de esta proteína, pero en ratones que carecen del gen (knock-out) se ha reportado esterilidad en los machos; aunque no se ha encontrado que su expresión afecte el desarrollo de las enfermedades priónicas se ha asociado con el desarrollo de ataxia y en los ratones carentes de la PrP produce sobreexpresión de PrPLP (14, 15).
ESPECTRO PATOLÓGICO
Las enfermedades producidas por agentes que cumplen los parámetros para ser llamados priones, conforman un grupo que ha aumentado progresivamente y se extiende a una amplia variedad de especies. En los humanos las enfermedades más ampliamente reconocidas como causadas por priones son las atribuidas a la proteína priónica PrP, codificada en el gen PRNP; éstas son: kuru, enfermedad de Creutzfeldt-Jakob clásica (ECJ), enfermedad de Creutzfeldt-Jakob nueva variante (nvECJ), síndrome de Gerstmann-Sträussler-Scheinker (GSS), insomnio fatal familiar (IFF), insomnio fatal esporádico (sIF), demencia atípica y paraparesia espástica con demencia. A su vez, con la ECJ pueden considerarse otras dos variantes que son la forma familiar (fECJ) y la forma infecciosa (iECJ) (10, 12).
En humanos existen otras enfermedades en las cuales el agente causal parece cumplir los parámetros para ser considerado un prión, como es el caso de la enfermedad de Alpers, caracterizada por crisis convulsivas, deterioro psicomotor y enfermedad hepática; es producida por una mutación en el gen de la ADN polimerasa mitocondrial γ (POLG) y se ha demostrado que puede ser transmitida (16, 17). Otra patología donde se han hallado indicios de que el agente etiológico posee un comportamiento similar a los priones es en la enfermedad de Köhlmeier-Degos, también llamada papulosis atrófica maligna, de esta enfermedad aún no se conoce su causa y se caracteriza por lesiones de tipo vascular en la piel y que también afectan el sistema nervioso central y el tracto gastrointestinal (18, 19).
Algunos autores han propuesto otras patologías en las cuales el agente etiológico puede comportarse como un prión, tal es el caso de la enfermedad de Alzheimer, cuya etiología es atribuida al beta amiloide que posee características comunes a la proteína priónica en su agregabilidad, cambio conformacional y transmisibilidad (20-23). De igual forma, se han sugerido las enfermedades de Huntington y de Parkinson, en las que la huntingtina y el α-sinuclein poseen características iguales a las observadas en las enfermedades priónicas, llevando a suponer que el espectro de las enfermedades priónicas se extiende a diversas patologías que son de alta frecuencia en la población (24-27).
En animales las enfermedades por priones afectan a una gran variedad de especies; sin embargo, al igual que en los humanos, sólo se ha detectado una proteína responsable de este grupo de males, que corresponde a la misma proteína PrP vista en los humanos. Las enfermedades producidas por priones en animales son: scrapie clásico (afecta a ovejas y cabras), scrapie atípico, también llamada Nor98 (afecta a ovejas), encefalitis transmisible del visón (afecta a visones), enfermedad caquectizante crónica (afecta a venados y alces), encefalopatía espongiforme bovina y encefalopatía espongiforme amiloidea bovina (afecta al ganado bovino), encefalopatía espongiforme exótica de los ungulados (afecta al órix surafricano, al órix árabe, al gran kudu y a los antílopes africanos) y encefalopatía espongiforme felina (afecta a gatos, tigres albinos, pumas y chitas). Las enfermedades priónicas en animales tienen en general un origen común que viene del scrapie, puesto que al morir las ovejas afectadas en los campos eran devoradas por los visones y de esa forma éstos adquirieron la encefalitis transmisible del visón; las otras encefalopatías transmisibles se generaron a partir de las harinas usadas como suplemento alimenticio que se produjeron usando ovejas contaminadas con scrapie, las que provocaron la epidemia de encefalitis espongiforme bovina y además fueron usadas en zoológicos y en los hogares, produciendo los otros tipos de encefalopatías transmisibles mencionadas (28-30).
En organismos como las levaduras, específicamente en el Saccharomyces cerevisiae, se ha detectado un importante número de proteínas que cumplen los parámetros para ser consideradas priones. Hasta el momento se han reportado: Ure2 (su gen es el URE3), Sup35 (su gen es el PSI+), RnqI (su gen es el PIN+), Sui (su gen es el SWI+), McaI (su gen es el MCA), Cyc8 (su gen es el OCT+) y el Mot3 (su gen es el MOT3+) (31, 32). También se han reportado priones en hongos, como es el caso de la Podospora anserina, en que se ha reportado el prión HET-s, el cual es producto del gen Het-s (33, 34). En el caso de los priones en levaduras y hongos, el efecto de éstos se refleja en la incapacidad del organismo para adaptarse a determinados nichos o la alteración de determinada vía metabólica. En la actualidad el estudio de priones en levaduras es una herramienta y un modelo muy importante para el conocimiento de la biología molecular de los priones en general (35, 36).
CLÍNICA DE LAS ENFERMEDADES PRIÓNICAS EN HUMANOS
Las enfermedades producidas por priones que afectan a los humanos son muy poco comunes, de la ECJ se presenta al año aproximadamente un caso por cada millón de habitantes, mientras que el GSS y las formas familiares de ECJ son aproximadamente 10 veces menos frecuentes, limitándose a pocos grupos familiares identificados en varias partes del mundo (37).
En el caso del kuru, se trata de una enfermedad limitada a un grupo poblacional localizado en las tierras altas de Nueva Guinea. Se pudo determinar que la enfermedad se producía por el consumo de carne humana contaminada con la enfermedad; al prohibir el canibalismo en la isla desde la década de los cincuenta, el número de casos ha disminuido, hasta considerarse cerca su erradicación. El descubrimiento de unos pocos casos en años recientes demuestra tiempos de incubación de esta enfermedad mayores a los 50 años, y se han señalado 5 años como el tiempo más corto de incubación (desde la ingesta del material contaminado al desarrollo de la enfermedad), con una media del tiempo de incubación estimada en 12 años. Se cree que el origen de la enfermedad fue por un caso de ECJ en la comunidad que fue consumido por los familiares y de esta forma se inició su propagación. El kuru afectó especialmente a mujeres y niños; posee un cuadro clínico que se ha dividido en tres estados, llamados: ambulante, sedentario y postración (que frecuentemente tiene un estado terminal prolongado). La enfermedad se inicia con dolor de cabeza o articular, durante un periodo variable, antes del inicio de ataxia cerebelosa. El trastorno cerebeloso es progresivo y el paciente pasa a un estado de dependencia; la demencia sólo se presenta en los estados tardíos de la enfermedad. La histopatología del kuru se caracteriza por pérdida de neuronas, vacuolización, hipertrofia y proliferación astrocítica, espongiosis, ausencia de inflamación y en algunos casos, placas amiloideas; los cambios se encuentran en el cerebro y la médula espinal, pero son más marcados en el cerebelo (38-41).
La ECJ puede encontrarse con una gran variedad fenotípica, dependiendo de su origen puede tratarse de una variante esporádica (a la cual pertenecen aproximadamente el 85% de los casos), de una forma familiar (poseen un tipo de herencia autosómica dominante) o de forma adquirida (cerca del 5% de los casos). En general la clínica de la ECJ consiste en una demencia rápidamente progresiva, acompañada de mioclonías y ataxia cerebelosa; la media para la edad de inicio son los 60 años, con un rango de edad entre los 45 y los 75 años; en promedio la enfermedad dura de 6 a 7 meses y el 90% de los pacientes fallecen durante el primer año de la enfermedad. Se han logrado determinar algunos factores relacionados con un tiempo mayor de sobrevida en los pacientes, como edades tempranas de inicio de la enfermedad, sexo femenino, ser heterocigótico para el codón 129, presencia de la proteína 14-3-3 en el líquido cefalorraquídeo, y poseer una cepa priónica tipo 2a (determinada por el patrón de glicosilación) (42). Se puede presentar como una de las seis variantes clínicas identificadas: clásica, Heidenhain, cerebelosa, panencefálica, amiotrófica y atípica. La forma clásica de ECJ se observa aproximadamente en el 60% de los casos y en algunos pacientes se describe una fase prodrómica de la enfermedad, consistente en fatiga, malestar, trastornos del sueño y alimentarios; se ha señalado que el primer síntoma puede ser uno de los siguientes: pérdida de memoria, dificultades para la marcha, vértigo o mareo, irritabilidad o agitación y parestesias (43). El deterioro en los pacientes se puede ver de una semana a otra; en los pacientes se aprecia rigidez, temblores, coreoatetosis, mioclonías y síntomas visuales como diplopía o parálisis supranucleares (44). La variante cerebelosa o atáxica se halla en aproximadamente el 10% de los pacientes, los cuales desarrollan la demencia antes que los trastornos motores y presentan mayor número de síntomas cognitivos y mioclonías. La variante de Heidenhain ocurre aproximadamente en el 20% de los casos y se caracteriza por presentar inicialmente trastornos visuales como alteraciones del campo visual, visión borrosa, percepción anómala de los colores y las formas, alucinaciones visuales, hemianopsia homónima y ceguera cortical; estos síntomas se relacionan con una demencia rápidamente progresiva y con los síntomas observados en la variante clásica.
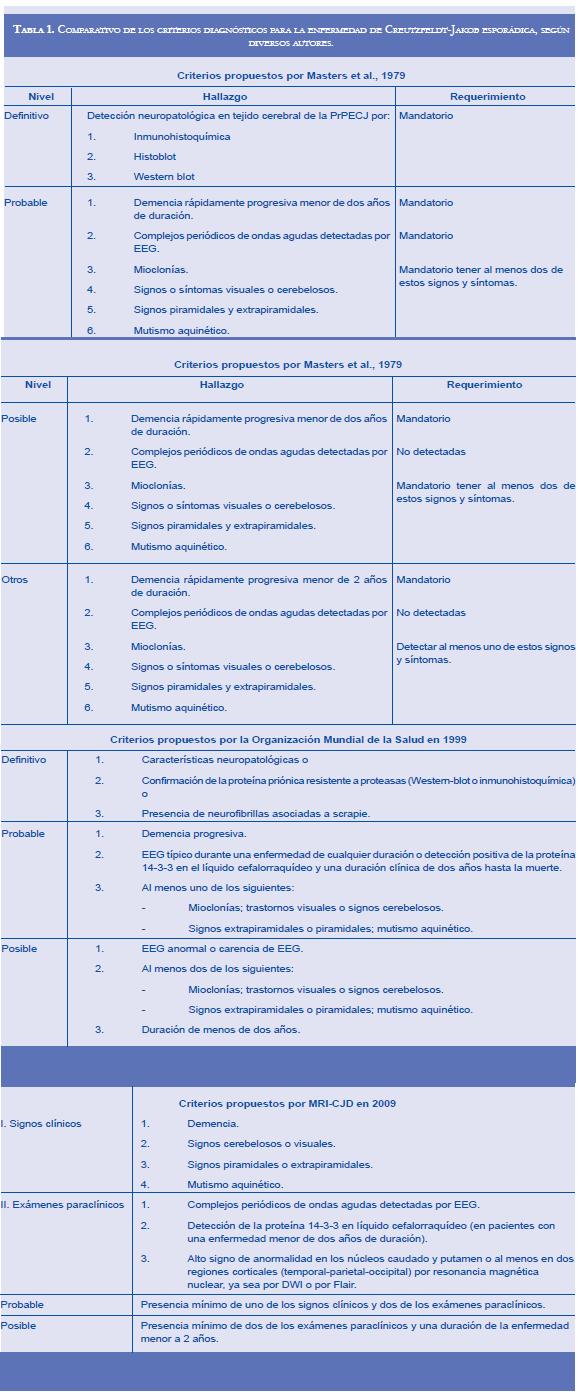
La variante panencefálica se encuentra especialmente en pacientes japoneses, notándose alteración tanto de sustancia gris como de sustancia blanca, con sintomatología tanto cortical como cerebelosa. La variante amiotrófica se caracteriza por iniciar con debilidad muscular progresiva, y en ella pueden aparecer fasciculaciones y neurodegeneración de la médula espinal. La variante atípica se observa en un 5 a 10% de los casos, puede durar más de dos años y afecta especialmente a individuos jóvenes; las mioclonías son menos frecuentes, el electroencefalograma (EEG) puede ser normal e inicia con alteración cognitiva y trastornos de comportamiento (45). Para el diagnóstico de la ECJ esporádica se han propuesto diversos parámetros, que han cambiado con el tiempo y probablemente seguirán modificándose en el futuro (ver comparativo de criterios diagnósticos en la tabla 1) (46-48). La histopatología de los casos esporádicos de ECJ reporta cambios espongiformes de corteza cerebral, ganglios basales, tálamo, corteza cerebelosa y el subiculum, además se observa pérdida neuronal, hipertrofia e hiperplasia de astrocitos y microglia, y abundantes agregados de PrP (depósitos amiloideos con diferentes patrones de agregación) (49, 50).
La forma adquirida de ECJ puede deberse al consumo de alimentos contaminados con priones, como es el caso del kuru y de la nueva variante (nvECJ), o serlo de forma iatrogénica. Se han reportado casos de transmisión iatrogénica de la ECJ por implantes de duramadre, personas sometidas a neurocirugía o a trasplante de córnea, a quienes se les ha realizado EEG estereotáctico (por electrodos contaminados), recibiendo gonadotropina y hormona de crecimiento humanas procedente de donantes cadavéricos. Aunque los priones son transmisibles, no son contagiosos; personas como los cuidadores o compañeros de los afectados no están en riesgo; no se transmite por contacto sexual, ni hay evidencia de transmisión materna, ni por leche materna; para el personal médico el principal riesgo se encuentra en la inoculación accidental de tejido infectado o fluidos; las transfusiones sanguíneas o la aplicación de componentes sanguíneos puede llevar a la transmisión de la enfermedad; también se considera que los procedimientos dentales pueden ser una forma de transmisión de la enfermedad. Es importante señalar que los priones pueden resistir los métodos convencionales de esterilización (37, 51-53). En las formas infecciosas de ECJ la presentación clínica cambia de acuerdo a la vía por la cual se adquirió la enfermedad; cuando la inoculación es directamente en el cerebro los periodos de incubación son de 16 a 28 meses y se presenta la variante panencefálica de la enfermedad, mientras que si la exposición es por vía periférica el tiempo de incubación puede durar de 4 a 30 años y la presentación clínica es una ataxia cerebelosa progresiva seguida de demencia (8, 45, 54). La Organización Mundial de la Salud estableció dos criterios diagnósticos para las formas iatrogénicas de la ECJ: síndrome cerebelar progresivo en un receptor de hormona de pituitaria humana derivada de cadáver, o el caso de la ECJ esporádica con exposición a riesgo reconocido (47). En las formas iatrogénicas de la ECJ los hallazgos neuropatológicos son variables; lo común es encontrar atrofia cerebral visible macroscópicamente, espongiosis y lesiones que se encuentran especialmente afectando el neoestriado, el puente y el cerebelo (más alterada la capa granular que la de células de Purkinje o la capa molecular) (55).
La nvECJ es producida por el consumo de carne vacuna contaminada por priones o por transfusión de sangre contaminada; hasta la fecha todos los casos de nvECJ han sido homocigóticos para metionina en el codón 129 (M/M) del gen PRNP, significando que para sufrir la enfermedad se debe consumir carne procedente de reses afectadas con encefalitis espongiforme bovina (EEB) y además poseer predisposición genética para sufrir la enfermedad; no obstante, también es posible que el tiempo de incubación para quienes portan un genotipo diferente al 129 M/M sea más prolongado y aún no se hayan reportado casos con genotipos distintos. La gran mayoría de los casos han sido reportados en Reino Unido, pero también han ocurrido en Francia, Irlanda, Italia, Holanda, España, Estados Unidos, Canadá y Japón (56-60). La enfermedad se presenta en individuos con un promedio de edad de 29 años (para un rango de edad de 12 a 74 años); desde el consumo del alimento contaminado hasta la aparición de la enfermedad pasa un tiempo de 15 a 20 años, y la enfermedad tiene una duración aproximada de 15 a 25 meses (37, 61, 62). Clínicamente la nvECJ se caracteriza por predominio de alteraciones conductuales y psiquiáticas y en algunos casos hay marcados trastornos sensoriales (disestesias o dolor en extremidades o cara); frecuentemente la queja inicial es de tipo psiquiátrico, siendo más común la depresión, pero también son frecuentes la ansiedad, el retraimiento y los cambios de comportamiento. Otros signos y síntomas observados son; delusiones, labilidad emocional, agresividad, insomnio, disartria y alucinaciones visuales y auditivas; con frecuencia muchos pacientes desarrollan un síndrome cerebeloso progresivo con ataxia para la marcha y en extremidades; la demencia usualmente se desarrolla en la fase tardía de la enfermedad; los miclonus son frecuentes en los pacientes y en ocasiones están precedidos de corea. Entre los cambios neuropsicológicos observados se presenta un deterioro cognitivo moderado a severo, con afectación de la memoria visual y verbal, disfunción ejecutiva y pérdida de habilidad nominal; el deterioro perceptual es menos frecuente (1, 63, 64). Para el diagnóstico de la nvECJ se han propuesto diversos parámetros que recolectan diferentes criterios diagnósticos (ver comparativo de criterios diagnósticos para nvECJ en la tabla 2), siendo la nvECJ una enfermedad de notificación obligatoria que requiere su respectiva vigilancia epidemiológica (47, 65). Histopatológicamente se diagnostica al encontrar en corteza cerebral y corteza cerebelar múltiples placas floridas por la tinción de hematoxilina eosina, en tinción para PrP se observan numerosos acúmulos de placas pequeñas y acúmulos amorfos de PrP pericelulares y perivasculares; en el estriado se aprecian cambios espongiformes severos y acumulación de PrP axonal y perineuronal; en tálamo y diencéfalo se ve marcada astrocitosis con pérdida neuronal; en tallo cerebral y médula espinal se evidencia acumulación de PrP en forma reticular y perineuronal que afecta la materia gris (47, 66).
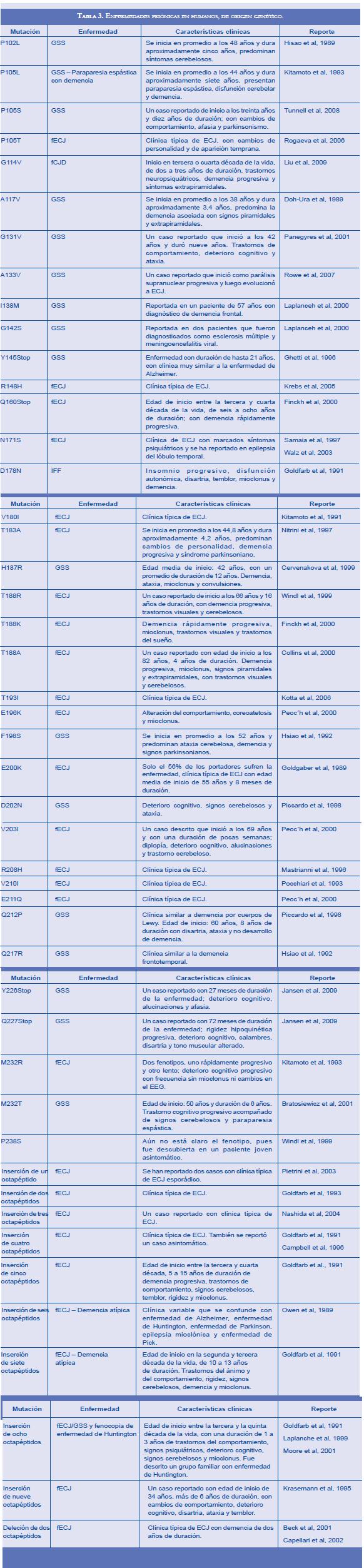
Las formas familiares de las enfermedades priónicas son unas verdaderas rarezas que se limitan a unos pocos grupos familiares dispersos por el mundo. Estas enfermedades se producen por mutaciones en el gen PRNP y se han descrito: la forma familiar de la enfermedad de Creutzfeldt-Jakob (fECJ), el síndrome de Gerstmann-Sträussler-Scheinker (GSS), el insomnio fatal familiar (IFF), la demencia atípica y la paraparesia espástica con demencia. Las diferentes mutaciones en el gen PRNP generan amplia variabilidad fenotípica (Tabla 3, donde se recopilan las diferentes mutaciones reportadas de PRNP que llevan al desarrollo de enfermedad priónica) (67-80). Aunque las mutaciones en PRNP generan un patrón de herencia autosómico dominante, sólo se ha detectado un caso aislado sin otros miembros de la familia afectados, por lo cual en algunos eventos se habla de GSS esporádico (81). En las formas genéticas de enfermedades por priones la paraparesia espástica con demencia es causada por la mutación P105L, identificada también como una de las variantes del GSS; igualmente, la demencia atípica es identificada como una forma familiar de ECJ que se produce por una inserción de seis o siete octapéptidos adicionales en la región de la proteína donde normalmente se encuentran cinco repeticiones en forma natural. En general el GSS se caracteriza por ataxia cerebelosa crónica, con signos piramidales y posteriormente demencia, y una evolución clínica más prolongada que la ECJ; sin embargo, la variabilidad fenotípica entre las diferentes mutaciones que generan GSS es bastante amplia. Igualmente, la fECJ posee amplia variabilidad fenotípica, como ocurre con el GSS, pero en general su clínica es muy similar a los casos esporádicos de ECJ y sólo se diferencian por la agregación familiar o por la presencia de una mutación en el gen PRNP (1, 54). En el diagnóstico histopatológico de fECJ básicamente se encuentran los mismos hallazgos presentes en las formas esporádicas de ECJ; en el GSS se halla encefalomielopatía con placas priónicas multicéntricas, degeneración talámica y cambios espongiformes variables en el cerebro (47). El IFF se desarrolla cuando hay sustitución de asparagina por ácido aspártico en el codón 179 (D178N) y adicionalmente los pacientes deben ser portadores de metionina en el codón 129; pacientes con la misma mutación (D178N) pero portadores de valina en el codón 129, desarrollan un fenotipo diferente, consistente en un fECJ. La mutación D178N parece tener origen ancestral y su distribución quizás se deba a un efecto fundador; a la fecha se han reportado casos de IFF en Italia, Alemania, Austria, España, Reino Unido, Francia, Finlandia, Australia, Estados Unidos, Japón, China y Marruecos (82-86). El IFF se inicia entre los 36 y los 62 años (51 años de promedio); la duración de la enfermedad es de 8 a 72 meses (18 meses en promedio); posee dos presentaciones clínicas: una de progresión rápida, con un promedio de duración de 9 meses y en la cual predominan los trastornos del sueño y la disautonomía, y otra de progresión lenta, con un promedio de duración de 30 meses, en la que predominan los trastornos motores. Normalmente la enfermedad se inicia con trastorno del sueño o cambio de personalidad; frecuentemente se presenta fatiga visual, diplopía, alteración del simpático, insomnio, somnolencia durante el día, ensoñaciones con sacudidas que semejan el contenido de los sueños "estupor onírico", trastornos motores y estado confusional progresivo (87). Los hallazgos neuropatológicos que caracterizan al IFF son atrofia selectiva de los núcleos talámicos ventral anterior y dorsomediano, con pérdida neuronal y gliosis masiva, también hay daños en las olivas inferiores y espongiosis en la corteza entorrinal y el neocortex (55).
Desde 1999 se han descrito unos pocos casos con clínica similar al IFF y que no pueden ser diferenciados neuropatológicamente del IFF, pero carecen de historia familiar de la enfermedad, no poseen mutaciones en el gen PRNP y todos los casos reportados son homocigóticos para metionina en el codón 129; esta enfermedad corresponde al llamado insomnio fatal esporádico sIF (88).
EXÁMENES PARACLÍNICOS
En las enfermedades priónicas se han usado los exámenes paraclínicos como ayudas para mejorar el diagnóstico, sin embargo el diagnóstico definitivo aún se determina por el estudio neuropatológico. Entre las pruebas realizadas se encuentran los exámenes imagenológicos, el examen de líquido cefalorraquídeo, el EEG, el estudio genético para el gen PRNP y la detección de proteínas específicas para la enfermedad como la proteína 14-3-3 y la proteína S100. Estos exámenes, y otros como hemograma (en enfermedades priónicas no muestra alteraciones) o imagenología de tórax y abdomen (por posibles neoplasias), son importantes para descartar diagnósticos diferenciales (89).
En el líquido cefalorraquídeo usualmente no se aprecian alteraciones en sus componentes como celularidad, glucosa, iones y proteínas (pueden estar levemente aumentadas); no obstante la detección de proteínas en el líquido cefalorraquídeo como la 14-3-3, S100β, tau, y la enolasa específica de neuronas (NSE), se consideran biomarcadores importantes para las enfermedades priónicas. Los niveles elevados de la proteína 14-3-3 ofrecen sensibilidad para el diagnóstico de ECJ de 95% y una especificidad del 93%, por lo cual la presencia de 14-3-3 en un paciente con demencia rápidamente progresiva se puede considerar confirmatoria del diagnóstico de ECJ; mas debe tenerse en cuenta que esta proteína también puede elevar sus niveles en pacientes con encefalitis virales, en hemorragias o infartos cerebrales recientes y en síndrome paraneoplásico. La proteína 14-3-3 que incluida por la OMS como criterio diagnóstico para la sECJ. Existen siete isotipos diferentes de 14-3-3, usualmente es detectada por Western-blot o por ELISA, se encuentra en una gran variedad de células, pero las neuronas poseen altos niveles. Los niveles elevados en el líquido cefalorraquídeo indican una extensa y rápida destrucción del tejido cerebral. Cuando se logra obtener determinaciones cuantitativas de 14-3-3 se ha encontrado que los niveles más altos son compatibles con ECJ rápidamente progresiva, sin embargo los niveles de 14-3-3 también se elevan en las formas familiares de ECJ y en algunos casos en GSS e IFF (10 a 13%, respectivamente) (90-95). La S100 es una proteína ácida de unión al calcio, un heterodímero conformado por dos subunidades: la alfa (α) y la beta (β); el isómeroβ (S100β) se halla en las células gliales. Las S100 son una familia de proteínas que comprende cerca de veinte proteínas diferentes y que se han relacionado con diferentes enfermedades neurodegenerativas; para el caso de ECJ, se ha establecido una correlación positiva de los niveles de S100β y el grado de espongiosis en el tálamo y el grado de gliosis de los ganglios basales y el cerebelo (96, 97). Los estudios han revelado que los niveles elevados de S100β en líquido cefalorraquídeo posee grados variables de sensibilidad y especificidad para el diagnóstico de ECJ, estos valores oscilan entre un 84% de sensibilidad y 90% de especificidad. Al igual que con la proteína 14-3-3, la S100β se encuentra en concentraciones menores en el líquido cefalorraquídeo de pacientes con GSS o IFF (89, 92). Otra de las proteínas en líquido cefalorraquídeo usadas como biomarcador para ECJ es la tau, la cual interviene en la patogénesis de diversas enfermedades neurodegenerativas al acumularse en las neuronas y formar ovillos neurofibrilares, en ECJ tau es profusamente liberada en el líquido cefalorraquídeo, indicando daño neuronal rápido (98). Se ha encontrado que los niveles de tau en el líquido cefalorraquídeo son menores en pacientes que presentan ECJ de larga duración, también se hallan niveles bajos al inicio o al final de la enfermedad; las concentraciones altas de tau se correlacionan con el grado de gliosis en los ganglios basales y cerebelo, junto con los cambios espongiformes. Niveles de tau superiores a 1.300 pg/ml se observan en las formas esporádicas y genéticas de ECJ, pero sólo se encuentran en aproximadamente el 40% de los casos de GSS y muy pocas veces en IFF; algunos autores le dan a la tau significancia para el diagnóstico de ECJ muy similar a la observada con la proteína 14-3-3, reportando una sensibilidad del 94% y una especificidad del 90% (92, 96, 99). Adicionalmente, se ha estudiado otra proteína en el líquido cefalorraquídeo, la enolasa específica de neuronas (NSE); una enzima que interviene en la vía glicolítica; al igual que con las otras proteínas, la NSE se eleva en el líquido cefalorraquídeo, denotando una pérdida rápida de neuronas; se ha evidenciado correlación entre los niveles elevados de NSE en líquido cefalorraquídeo y el grado de gliosis en ganglios basales y cerebelo, junto con los cambios espongiformes en el tálamo. Algunos autores dan un 78% de sensiblidad para el diagnóstico de ECJ cuando se encuentran niveles de NSE superiores a los 35 ng/ml en líquido cefalorraquídeo; otros autores han reportado que en las formas familiares de ECJ tuvo una sensibilidad del 64% y no fue detectable en casos de GSS e IFF. Se han propuesto diferentes proteínas como marcadores en líquido cefalorraquídeo para el diagnóstico de las enfermedades priónicas, como los niveles de ApoE, de beta amiloide o de PrP; sin embargo, aún no hay resultados concluyentes al respecto y sólo la proteína 14-3-3 fue incluida como criterio diagnóstico para la ECJ (89, 92, 96, 100, 101).
Herramientas diagnósticas como el electroencefalograma (EEG) hacen parte de los criterios diagnósticos en las enfermedades priónicas; con el progreso de la enfermedad y el deterioro constante se espera encontrar un EEG alterado por actividad de ondas lentas, la actividad caracteristica de las enfermedades priónicas son los complejos de ondas agudas periódicas de uno o dos hertz. El EEG posee una sensibilidad del 65% y una especificidad del 86% en el diagnóstico; puede que los cambios característicos de la enfermedad no se encuentren en un primer EEG, por lo cual es aconsejable repetir el EEG cada semana. Los cambios que caracterizan a la ECJ también se han reportado en enfermedad de Alzheimer, encefalopatía hepática, toxicidad por drogas y trastornos metabólicos. Aunque el EEG es considerado como característico de la sECJ, en las formas familiares como en el GSS usualmente no se observan los cambios característicos de las enfermedades priónicas. En las fases iniciales de la enfermedad el EEG exhibe una lentificación difusa de la actividad de fondo, con ritmos delta y theta; al avanzar la enfermedad pueden verse algunas descargas periódicas aisladas y esporádicas. Con el progreso de la enfermedad el EEG cambia a un patrón conocido, FIRDA (frontal intermittent rhytmic delta activity), con ondas delta de voltaje elevado (100-150 mV); al continuar progresando la enfermedad se aprecia el patrón típico de la enfermedad, con actividad periódica, sincrónica y simétrica, con grafoelementos bi o trifásicos de aproximadamente 100 a 300 milisegundos de duración, que se repiten en un intervalo de 0,7 a 1,5 segundos. Ya en estado muy avanzado de la enfermedad en el EEG los complejos periódicos son sustituidos por actividad theta lenta con marcada ritmicidad y voltaje entre medio y alto. En la fase terminal el EEG revela desaparición de los complejos periódicos y aplanamiento de la actividad de fondo (56, 81, 89, 102, 103).
Los estudios neuroimagenológicos han sido considerados como criterio diagnóstico en las enfermedades priónicas, con una sensibilidad de 67% y una especificidad del 93% (la sensibilidad y la especificidad han variado con diferentes autores); si bien algunos autores consideran exámenes como la resonancia magnética (RM) una prueba con poca sensibilidad, estudios recientes le atribuyen alta sensibilidad y especificidad moderada para el diagnóstico de ECJ; además, es importante para descartar otras patologías que presentan deterioro neurológico similar (48, 93, 104, 105). En la RM los cambios que normalmente se observan en ECJ son hiperintensidades en las secuencias T2 de los ganglios basales (generalmente en caudado y putamen) y en el área cortical, especialmente en las áreas occipitales y temporales; con el uso de secuencias como la DWI (diffusion-weight imaging) y la FLAIR (fast fluid-attenuated inversión recovery), las alteraciones en ganglios basales y en corteza son más fácilmente identificables, mejorando notoriamente la sensibilidad y especificidad de la técnica. Usualmente la tomografía axial computarizada (TAC) es normal, aunque el paciente se encuentre muy sintomático, o puede también mostrar anormalidades inespecíficas como atrofia generalizada. Con el uso de SPECT se ha encontrado hipoperfusión cerebral especialmente en los lóbulos occipitales, cerebelo o en un hemisferio completo, el patrón de hipoperfusión puede ser variable. En tomografía por emisión de positrones (PET) se ha reportado hipometabolismo asimétrico en varias áreas corticales en los pacientes con ECJ. La combinación de RMN con espectroscopia tiene prometedores resultados, puesto que un estudio reciente halló que la razón de N-acetil-aspartato a creatina para pacientes con ECJ presentan niveles menores o iguales a 1,21 para un 100% de sensibilidad y de valor predictor negativo, mientras que se determinaron iguales características de sensibilidad y valor predictor negativo en pacientes que no tienen ECJ para la razón mayor de 1,05 de N-acetil-aspartato a mio-inositol (106-112).
Estudios genéticos como el de la secuencia del gen pueden realizarse para identificar una mutación específica causante de la enfermedad, en este caso se debe recordar que cerca del 90% de los casos corresponden a formas esporádicas donde no hay mutaciones en el gen; además, éstas poseen un patrón de herencia autosómico dominante, por lo que se debe esperar una historia familiar importante relacionada con la enfermedad; no obstante, algunas mutaciones en el gen PRNP han sido identificadas en un solo individuo y en pacientes a los cuales no se les ha detectado historia familiar de enfermedad por priones. Otro de los estudios genéticos que se realizan en las enfermedades priónicas es la identificación del polimorfismo 129, que puede presentar una variación de metionina (M) o valina (V) en dicha posición. Se considera que los individuos homocigóticos para metionina (M/M) sufren mayor riesgo de desarrollar enfermedades priónicas; aproximadamente el 71% de los casos de ECJ esporádico en Francia poseen el genotipo M/M (en Reino Unido representan el 68% de los casos), mientras que la frecuencia del genotipo M/M en la población francesa es del 41% (en Reino Unido el 39% de la población es M/M); a su vez el genotipo M/V se considera como un factor protector, pues en el Reino Unido sólo el 16% de los afectados de sECJ son M/V y el 50% de la población del Reino Unido es M/V. El polimorfismo 129 también es un modificador importante de la clínica de los pacientes afectados, encontrando las formas más agresivas de la enfermedad en quienes portan el genotipo M/M, y las formas más crónicas de la enfermedad en quienes portan el genotipo M/V. No solamente se observan cambios en la presentación clínica de la enfermedad con diferencias en el polimorfismo 129, también se ha encontrado que los resultados de exámenes como la RMN, el EEG y la proteína 14-3-3 pueden variar dependiendo de este genotipo (48, 105, 113-115).
TRATAMIENTOS
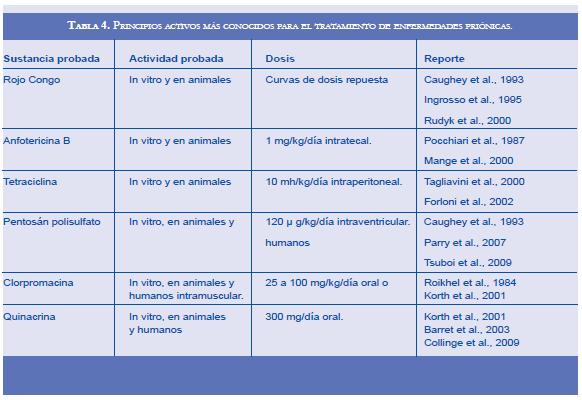
A pesar de haberse probado una gran cantidad de principios activos con efecto antipriónico, a la fecha no hay un tratamiento que cambie significativamente el cuadro clínico de las enfermedades por priones, lo único que se ofrece a los pacientes son cuidados paliativos. Algunas de las sustancias probadas poseen grados variables de efectividad para combatir las enfermedades priónicas (Tabla 4). Sustancias como los compuestos polianiónicos, de los cuales hacen parte los glicosaminoglicanos, entre los que se encuentran el heparán sulfato, la heparina, el dermatán sulfato, el keratán sulfato, el condroitín sulfato o análogos semisintéticos como el pentosán polisulfato y el dextrán sulfato, tienen la capacidad de unirse a PrP y competir con la transformación de PrPC en PrPSc (116-119). De estas sustancias el pentosán polisulfato es un anticoagulante y antiinflamatorio usado en veterinaria, y ya ha sido probado en varios casos humanos, reportando como resultado un aumento en la sobrevida de los pacientes sin detener la progresión de la enfermedad; en estudios comparativos con otras sustancias antipriónicas usadas como quinacrina y anfotericina B se ha recomendado al pentosán polisulfato como el compuesto con mejor resultados al tratamiento. Básicamente esta molécula actúa como un inhibidor competitivo con la PrP, al unirse a la PrPC y a la PrPSc (120-123). El rojo Congo es otra molécula que se une a la PrP, pero en el caso del rojo Congo se une a las formas fibrilares impidiendo la polimerización de las fibrillas y de esta forma detiene la agregación; es poco probable que el rojo Congo pueda ser usado en tratamiento para humanos debido a su alta toxicidad, pero se está trabajando en derivados de esta molécula que pueden tener mejores posibilidades en la terapéutica de enfermedades de depósitos amiloideos como las enfermedades priónicas (124-128).
A la anfotericina B se le ha encontrado que tiene la capacidad de prolongar el tiempo de incubación de los priones; sin embargo, al manifestarse la enfermedad no se le vio efecto en los animales tratados. Al igual que la mayoría de los fármacos usados en tratamientos experimentales contra los priones, no está muy claro el mecanismo de cómo actúa la anfotericina B; se cree que ella afecta los microdominios lipídicos donde se halla la proteína priónica, lo que altera las interacciones de ésta e impide la transformación en la forma patológica. Debido a la toxicidad de la anfotericina B se han desarrollado derivados, como la MS-8209, que por ser menos tóxica se puede usar en dosis más alta y ha mostrado resultados un poco mejores que la anfotericina B (129-136).
La quinacrina, un medicamento usado tradicionalmente en el tratamiento de la malaria, tiene actividad antipriónica en cultivos celulares y con resultados muy discutidos en animales y humanos; no es claro el mecanismo de acción antipriónico de la quinacrina, pero se sabe que interactúa con la proteína priónica, mas no afecta la capacidad de agregación ni la resistencia de las formas fibrilares de los priones, aunque inhibe la formación de agregados priónicos posiblemente debido a que puede afectar la vía lisosomal y de esta forma altera el balance de síntesis y catabolismo de la proteína priónica. El estudio más amplio realizado a la fecha, en el cual se incluyeron 107 pacientes con ECJ para evaluar la seguridad y eficiencia de la quinacrina usando una dosis inicial de 1 gr repartido en tomas cada 6 horas y seguido por 100 mg cada 8 horas, reportó que la medicación fue tolerada por los pacientes, aunque el curso clínico de la enfermedad no se afectó significativamente. Algunos autores atribuyen las fallas en el tratamiento con quinacrina a bajas concentraciones tisulares del medicamento, posiblemente relacionado con la eficiencia que la quinacrina posee para atravesar la barrera hematoencefálica. No obstante, se ha detectado que la quinacrina se acumula en el cerebro y parece presentarse resistencia de los priones a esta droga (122, 137-142).
Otro antibiótico que ha mostrado actividad antipriónica es la tetraciclina, la cual parece interactuar con la cadena de aminoácidos de la proteína priónica en una región que es importante para la conversión en la forma patológica. Las tetraciclinas poseen algunas características que las convierten en un fármaco atractivo para el tratamiento de las enfermedades priónicas, como es el caso de poder atravesar la barrera hematoencefálica, reducir la capacidad infecciosa de los priones y hacerlos más sensibles a las proteasas, consideradas características fundamentales de los priones. Pese a ello, aún no se conocen estudios sobre el efecto de las tetraciclinas en el tratamiento de humanos afectados con ECJ (143-145).
La clorpromacina es un medicamento usado desde hace varios años como antipsicótico, hallándose experimentalmente que posee actividad antipriónica tanto in vitro como en modelos animales, especialmente en combinación con quinacrina; debido a que ambos atraviesan la barrera hematoencefálica, algunos autores consideraron la combinación de ambas como la mejor alternativa terapéutica para la ECJ. Hay pocos reportes de los efectos sobre los pacientes con enfermedad priónica tratados con clorpromacina tanto en monoterapia como combinada con quinacrina, generalmente en estos casos el tratamiento ha sido realizado para controlar síntomas psiquiátricos, y se ha reportado una actividad antipriónica de la clorpromacina mucho menor que la encontrada con la quinacrina. El mecanismo por medio del cual actúa la clorpromacina sobre los priones también es desconocido, posiblemente se debe al efecto de la clorpromacina sobre la vía dependiente de clatrina, en la cual se dan importantes interacciones de la proteína priónica (116, 142, 146-149).
Las estrategias farmacológicas para tratar las enfermedades priónicas se extienden a muchos otros principios activos, así como el uso de otras alternativas terapéuticas, como: anticuerpos contra la proteína priónica, la utilización de RNA de interferencia, e incluso la inmunización contra la proteína priónica patológica. Éste es un frente intenso de investigación en la actualidad y es posible que los resultados sean variables, teniendo en cuenta la gran variedad fenotípica de las enfermedades priónicas, pues ya se ha reportado que la actividad de medicamentos antipriónicos es dependiente de la cepa de prión que está siendo tratada. Sin embargo el conocimiento alcanzado sobre los priones y la gran cantidad de propuestas farmacológicas hacen probable el desarrollo de una terapéutica más efectiva contra las enfermedades priónicas en los próximos años (116, 150-153).
ALGUNOS ASPECTOS MOLECULARES DE LOS PRIONES
De acuerdo al polimorfismo metionina o valina en el codón 129 de la proteína priónica, se han identificado los genotipos M/M, M/V y V/V; y de acuerdo al patrón de migración electroforética de la proteína también se ha diferenciado a la proteína priónica patológica en dos tipos: el tipo 1, con un peso molecular de 21 kDa y un sitio de clivaje en el residuo 82, y el tipo 2 con un peso molecular de 19 kDa y un sitio de clivaje en el residuo 97. Con estas características moleculares se ha establecido una clasificación molecular de la enfermedad, en la cual se pueden presentar los subtipos MM1, MM2, MV1, MV2, VV1 y VV2. Identificar estas variantes moleculares de los priones ha permitido correlacionar la clínica de la enfermedad con el subtipo molecular que se presenta y por lo tanto se han establecido relaciones con la variabilidad fenotípica (114, 154-156).
Igualmente, debido al patrón de migración de la proteína priónica en la electroforesis se han identificado tres tipos de proteína priónica, de acuerdo con su grado de glicosilación: altamente glicosilada, levemente glicosilada y no glicosilada. Gracias a esta diferenciación se han logrado identificar diferentes cepas de proteína priónica y de esta forma se ha determinado cómo la proteína priónica pasa la barrera de especies, como en el caso de la nvECJ y la EEB. También ha servido para identificar las cepas según su origen, pues se ha encontrado que la glicosilación varía dependiendo del origen de la proteína priónica, de tal forma que la glicosilación es diferente en la proteína priónica dependiendo de si ésta es de origen genético, o si es infecciosa (157-159).
Adicionalmente al polimorfismo en el codón 129, en el gen de PRNP se han identificado un gran número de polimorfismos, algunos silenciosos y otros a los cuales no se les ha establecido claramente su función en la patogénesis de las enfermedades priónicas. Entre los rasgos estructurales de la proteína priónica se encuentra además el poseer un puente bisulfuro y dos sitios en la proteína que pueden ser glicosilados (113, 160). La PrPC está conformada por tres hélices alfa y un pequeño pliegue beta; luego, al sufrir la transformación a PrPSc cambia la proporción de pliegues beta, de 3% en la PrPC original, a un 45% aproximadamente en la PrPSc; es este cambio conformacional el que cambia las características físico-químicas de la proteína priónica, provocando que pase a ser una proteína más estable y difícil de degradar ante factores químicos y físicos; de éstos, la capacidad de resistir proteasas como la proteinasa K se convierte en uno de los rasgos principales que permiten identificar a la proteína patológica en los tejidos y es una de las técnicas que nos permiten dar un diagnóstico definitivo de las enfermedades priónicas. La PrPC también posee la capacidad de fijar entre dos a seis átomos de cobre en una porción donde se encuentran los octapéptidos, esta capacidad de fijar cobre la pierde la PrPSc y se cree que éste puede ser uno de los mecanismos por los cuales la PrPSc lleva a neurodegeneración, puesto que una probable función de la PrPC al captar el cobre sería la de actuar como antioxidante y esta capacidad se perdería al pasar a la forma fibrilar (1, 6, 161-163).
La función de la PrPC ha sido un tema de gran investigación, puesto que los ratones que carecen de este gen se desarrollan aparentemente normal, pero en los estudios realizados se han atribuido funciones a PrP tales como: factor de transcripción que interviene en la polaridad celular, regulación de glicosilación modulando la expresión neuronal de cobre, en transducción de señales, en memoria, en transmisión sináptica, ritmo circadiano, reacciones de inflamación, proliferación y diferenciación celular, entre otras. Probablemente el conocimiento de su función y sus interacciones moleculares, tanto de PrP como de la familia de proteínas a la que pertenece, ayudará a esclarecer la gran cantidad de dudas que rodean a estas enfermedades y allanará el camino de la terapia no solo para las enfermedades priónicas, sino también para un importante número de enfermedades neurodegenerativas (163-168).
Agradecimientos
Este trabajo fue realizado gracias a Colciencias y a la Universidad de Antioquia, por medio del proyecto "Identificación de marcadores de homeostasis celular en tejido cerebral de enfermedad de Alzheimer familiar", código: 1115-493-2613, financiado bajo el contrato 628-2009.
REFERENCIAS
1. COLLINGE J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24: 519-550.
2. NARANG H. A critical review of the nature of the spongiform encephalopathy agent: protein theory versus virus theory. Exp Biol Med (Maywood) 2002; 227(1): 4-19.
3. COCHEREAU I. Unconventional transmissible agents. J Fr Ophtalmol 2004; 27: 414-416.
4. LAGNADO J. Past times - From pabulum to prions (via DNA) a tale of two Griffiths. The Biochemist 2009:33-35.
5. COLLINGE J. Review. Lessons of kuru research: background to recent studies with some personal reflections. Philos Trans R Soc Lond B Biol Sci 2008; 363: 3689-3696.
6. COHEN FE, PRUSINER SB. Pathologic conformations of prion proteins. Annu Rev Biochem 1998; 67: 793-819.
7. MORANGE M. What history tells us VIII. The progressive construction of a mechanism for prion diseases. J Biosci 2007; 32: 223-227.
8. BELAY ED. Transmissible spongiform encephalopathies in humans. Annu Rev Microbiol 1999; 53: 283-314.
9. ESCUDERO-TORRELLA J. Chronology of the new variant of Creutzfeldt-Jakob disease. Rev Neurol 2000; 31: 141-147.
10. KOVACS GG, BUDKA H. Molecular pathology of human prion diseases. Int J Mol Sci 2009; 10: 976-999.
11. AGUZZI A, MONTRASIO F, KAESER PS. Prions: health scare and biological challenge. Nat Rev Mol Cell Biol 2001; 2: 118-126.
12. RYOU C. Prions and prion diseases: fundamentals and mechanistic details. J Microbiol Biotechnol 2007; 17: 1059-1070.
13. BERTRAM L, TANZI RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest 2005; 115: 1449-1457.
14. BEHRENS A, GENOUD N, NAUMANN H, RULICKE T, JANETT F, HEPPNER FL ET AL. Absence of the prion protein homologue Doppel causes male sterility. EMBO J 2002; 21: 3652-3658.
15. MOORE RC, LEE IY, SILVERMAN GL, HARRISON PM, STROME R, HEINRICH C, ET AL. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292: 797-817.
16. MANUELIDIS EE, RORKE LB. Transmission of Alpers' disease (chronic progressive encephalopathy) produces experimental Creutzfeldt-Jakob disease in hamsters. Neurology 1989; 39: 615-621.
17. NGUYEN KV, OSTERGAARD E, RAVN SH, BALSLEV T, DANIELSEN ER, VARDAG A ET AL. POLG mutations in Alpers syndrome. Neurology 2005; 65: 1493-1495.
18. PIERCE RN, SMITH GJ. Intrathoracic manifestations of Degos' disease (malignant atrophic papulosis). Chest 1978; 73: 79-84.
19. SLAVIERO F, ANNES RD, FRIGHETTO L, SCHIRMER LM, VANZIN JR, FROHLICH AC ET AL. Kohlmeier-Degos Disease (malignant atrophic papulosis) and neurologic involvement. Arq Neuropsiquiatr 2009; 67: 692-694.
20. CASTELLANI RJ, PERRY G, SMITH MA. Prion disease and Alzheimer's disease: pathogenic overlap. Acta Neurobiol Exp (Wars) 2004; 64: 11-17.
21. LUPI O, PERYASSU MA. An emerging concept of prion infections as a form of transmissible cerebral amyloidosis. Prion 2007; 1: 223-227.
22. MEYER-LUEHMANN M, COOMARASWAMY J, BOLMONT T, KAESER S, SCHAEFER C, KILGER E ET AL. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313: 1781-1784.
23. RIEK R. Cell biology: infectious Alzheimer's disease? Nature 2006; 444: 429-431.
24. LI JY, ENGLUND E, HOLTON JL, SOULET D, HAGELL P, LEES AJ ET AL. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 2008; 14: 501-503.
25. MERIIN AB, ZHANG X, HE X, NEWNAM GP, CHERNOFF YO, SHERMAN MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157: 997-1004.
26. MOORE RC, XIANG F, MONAGHAN J, HAN D, ZHANG Z, EDSTROM L ET AL. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet 2001; 69: 1385-1388.
27. REN PH, LAUCKNER JE, KACHIRSKAIA I, HEUSER JE, MELKI R, KOPITO RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 2009; 11: 219-225.
28. BENESTAD SL, ARSAC JN, GOLDMANN W, NOREMARK M. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res 2008; 39: 19.
29. STEVENS KB, DEL RV V, GUITIAN J. Classical sheep scrapie in Great Britain: spatial analysis and identification of environmental and farm-related risk factors. BMC Vet Res 2009: 5-33.
30. WATTS JC, BALACHANDRAN A, WESTAWAY D. The expanding universe of prion diseases. PLoS Pathog 2006; 2: e26.
31. WICKNER RB, EDSKES HK, SHEWMAKER F, KRYNDUSHKIN D, NEMECEK J. Prion variants, species barriers, generation and propagation. J Biol 2009; 8: 47.
32. SONDHEIMER N, LÓPEZ N, CRAIG EA, LINDQUIST S. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J 2001; 20: 2435-2442.
33. DALSTRA HJ, SWART K, DEBETS AJ, SAUPE SJ, HOEKSTRA RF. Sexual transmission of the [Het-S] prion leads to meiotic drive in Podospora anserina. Proc Natl Acad Sci U S A 2003; 100: 6616-6621.
34. DALSTRA HJ, VAN DER ZR, SWART K, HOEKSTRA RF, SAUPE SJ, DEBETS AJ. Non-mendelian inheritance of the HET-s prion or HET-s prion domains determines the het-S spore killing system in Podospora anserina. Fungal Genet Biol 2005; 42: 836-847.
35. SAUPE SJ, SUPATTAPONE S. What makes a good prion? Conference on Prion Biology. EMBO Rep 2006; 7: 254-258.
36. WICKNER RB, TAYLOR KL, EDSKES HK, MADDELEIN ML, MORIYAMA H, ROBERTS BT. Prions in Saccharomyces and Podospora spp.: protein-based inheritance. Microbiol Mol Biol Rev 1999; 63: 844-861.
37. COLLINGE J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry 2005; 76: 906-919.
38. ALPERS MP. Review. The epidemiology of kuru: monitoring the epidemic from its peak to its end. Philos Trans R Soc Lond B Biol Sci 2008; 363: 3707-3713.
39. COLLINGE J, WHITFIELD J, MCKINTOSH E, BECK J, MEAD S, THOMAS DJ ET AL. Kuru in the 21st century-an acquired human prion disease with very long incubation periods. Lancet 2006; 367: 2068-2074.
40. MCLEAN CA. Review. The neuropathology of kuru and variant Creutzfeldt-Jakob disease. Philos Trans R Soc Lond B Biol Sci 2008; 363: 3685-3687.
41. WADSWORTH JD, JOINER S, LINEHAN JM, ASANTE EA, BRANDNER S, COLLINGE J. Review. The origin of the prion agent of kuru: molecular and biological strain typing. Philos Trans R Soc Lond B Biol Sci 2008; 363: 3747-3753.
42. POCCHIARI M, PUOPOLO M, CROES EA, BUDKA H, GELPI E, COLLINS S ET AL. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 2004; 127: 2348-2359.
43. RABINOVICI GD, WANG PN, LEVIN J, COOK L, PRAVDIN M, DAVIS J ET AL. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology 2006; 66: 286-287.
44. ARMSTRONG RA. Creutzfeldt-Jakob disease and vision. Clin Exp Optom 2006; 89: 3-9.
45. ZIVKOVIC S, BOADA M, LÓPEZ O. Review of Creutzfeldt-Jakob disease and other prion diseases. Rev Neurol 2000; 31: 1171-1179.
46. WEBER T, OTTO M, BODEMER M, ZERR I. Diagnosis of Creutzfeldt-Jakob disease and related human spongiform encephalopathies. Biomed Pharmacother 1997; 51: 381-387.
47. World Health Organization, Department of communicable disease surveillance and response. WHO Recommended surveillance standards. Second edition 1999:1-116.
48. ZERR I, KALLENBERG K, SUMMERS DM, ROMERO C, TARATUTO A, HEINEMANN U ET AL. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132: 2659-2668.
49. ARIZA A. El patólogo ante las encefalopatías espongiformes transmisibles. Rev Esp Patol 2002; 35: 49-62.
50. KOVACS GG, HEAD MW, HEGYI I, BUNN TJ, FLICKER H, HAINFELLNER JA ET AL. Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol 2002; 12: 1-11.
51. MABBOTT N, TURNER M. Prions and the blood and immune systems. Haematologica 2005; 90: 542-548.
52. SZYMANSKA J. Microbiological risk factors in dentistry. Current status of knowledge. Ann Agric Environ Med 2005; 12: 157-163.
53. HOUSTON F, MCCUTCHEON S, GOLDMANN W, CHONG A, FOSTER J, SISO S ET AL. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood 2008; 112: 4739-4745.
54. GLATZEL M, STOECK K, SEEGER H, LUHRS T, AGUZZI A. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005; 62: 545-552.
55. MIKOL J. Neuropathology of prion diseases. Biomed Pharmacother 1999; 53: 19-26.
56. KNIGHT RS, WILL RG. Prion diseases. J Neurol Neurosurg Psychiatry 2004; 75 Suppl 1: i36-i42.
57. MALLUCCI G, COLLINGE J. Update on Creutzfeldt-Jakob disease. Curr Opin Neurol 2004; 17: 641-647.
58. SHINDE A, KUNIEDA T, KINOSHITA Y, WATE R, NAKANO S, ITO H ET AL. The first Japanese patient with variant Creutzfeldt-Jakob disease (vCJD). Neuropathology; 2009.
59. VOROU RM, PAPAVASSILIOU VG, TSIODRAS S. Emerging zoonoses and vector-borne infections affecting humans in Europe. Epidemiol Infect 2007; 135: 1231-1247.
60. DIETZ K, RADDATZ G, WALLIS J, MULLER N, ZERR I, DUERR HP ET AL. Blood transfusion and spread of variant Creutzfeldt-Jakob disease. Emerg Infect Dis 2007; 13: 89-96.
61. MENÉNDEZ-GONZÁLEZ M, GARCÍA-FER- NÁNDEZ C, SUÁREZ-SAN MARTÍN E, ANTÓN-GONZÁLEZ C, BLÁZQUEZ-MENES B. The chronopathology of prion encephalopathies. Rev Neurol 2004; 39: 962-965.
62. TREVITT CR, SINGH PN. Variant Creutzfeldt-Jakob disease: pathology, epidemiology, and public health implications. Am J Clin Nutr 2003; 78(3 Suppl): 651S-656S.
63. CORDERY RJ, ALNER K, CIPOLOTTI L, RON M, KENNEDY A, COLLINGE J ET AL. The neuropsychology of variant CJD: a comparative study with inherited and sporadic forms of prion disease. J Neurol Neurosurg Psychiatry 2005; 76: 330-336.
64. SPENCER MD, KNIGHT RS, WILL RG. First hundred cases of variant Creutzfeldt-Jakob disease: retrospective case note review of early psychiatric and neurological features. BMJ 2002; 324: 1479-1482.
65. WILL RG, ZEIDLER M, STEWART GE, MACLEOD MA, IRONSIDE JW, COUSENS SN ET AL. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000; 47: 575-582.
66. IRONSIDE JW, HEAD MW, BELL JE, MCCARDLE L, WILL RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology 2000; 37: 1-9.
67. KOTTA K, PASPALTSIS I, BOSTANTJOPOULOU S, LATSOUDIS H, PLAITAKIS A, KAZIS D ET AL. Novel mutation of the PRNP gene of a clinical CJD case. BMC Infect Dis 2006; 6: 169.
68. LIU Z, JIA L, PIAO Y, LU D, WANG F, LV H ET AL. Creutzfeldt-Jakob disease with PRNP G114V mutation in a Chinese family. Acta Neurol Scand 2009.
69. BEJOT Y, OSSEBY GV, CAILLIER M, MOREAU T, LAPLANCHE JL, GIROUD M. Rare E196K mutation in the PRNP gene of a patient exhibiting behavioral abnormalities. Clin Neurol Neurosurg 2009.
70. JANSEN C, PARCHI P, CAPELLARI S, VERMEIJ AJ, CORRADO P, BAAS F ET AL. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol 2009.
71. SHIGA Y, SATOH K, KITAMOTO T, KANNO S, NAKASHIMA I, SATO S ET AL. Two different clinical phenotypes of Creutzfeldt-Jakob disease with a M232R substitution. J Neurol 2007; 254: 1509-1517.
72. ROEBER S, KREBS B, NEUMANN M, WINDL O, ZERR I, GRASBON-FRODL EM ET AL. Creutzfeldt-Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17-kDa prion protein fragment. Acta Neuropathol 2005; 109: 443-448.
73. GOLDFARB LG, BROWN P, MCCOMBIE WR, GOLDGABER D, SWERGOLD GD, WILLS PR ET AL. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci U S A 1991; 88: 10926-10930.
74. KRASEMANN S, ZERR I, WEBER T, POSER S, KRETZSCHMAR H, HUNSMANN G ET AL. Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Brain Res Mol Brain Res 1995; 34: 173-176.
75. LAPLANCHE JL, HACHIMI KH, DURIEUX I, THUILLET P, DEFEBVRE L, DELASNERIE-LAUPRETRE N ET AL. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain 1999; 122 : 2375-2386.
76. PANEGYRES PK, TOUFEXIS K, KAKULAS BA, CERNEVAKOVA L, BROWN P, GHETTI B ET AL. A new PRNP mutation (G131V) associated with Gerstmann-Straussler-Scheinker disease. Arch Neurol 2001; 58: 1899-1902.
77. FINCKH U, MULLER-THOMSEN T, MANN U, EGGERS C, MARKSTEINER J, MEINS W ET AL. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 2000; 66: 110-117.
78. KREBS B, LEDERER RM, WINDL O, GRASBON-FRODL EM, ZERR I, KRETZSCHMAR HA. Creutzfeldt-Jakob disease associated with an R148H mutation of the prion protein gene. Neurogenetics 2005; 6: 97-100.
79. PEOC'H K, MANIVET P, BEAUDRY P, ATTANE F, BESSON G, HANNEQUIN D ET AL. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 2000; 15: 482.
80. BRATOSIEWICZ J, LIBERSKI PP, KULCZYCKI J, KORDEK R. Codon 129 polymorphism of the PRNP gene in normal Polish population and in Creutzfeldt-Jakob disease, and the search for new mutations in PRNP gene. Acta Neurobiol Exp (Wars) 2001; 61: 151-156.
81. BRANDEL JP. Clinical aspects of human spongiform encephalopathies, with the exception of iatrogenic forms. Biomed Pharmacother 1999; 53: 14-18.
82. BALDIN E, CAPELLARI S, PROVINI F, CORRADO P, LIGUORI R, PARCHI P ET AL. A case of fatal familial insomnia in Africa. J Neurol 2009; 256: 1778-1779.
83. MEDORI R, TRITSCHLER HJ. Prion protein gene analysis in three kindreds with fatal familial insomnia (FFI): codon 178 mutation and codon 129 polymorphism. Am J Hum Genet 1993; 53: 822-827.
84. ZARRANZ JJ, DIGON A, ATARES B, RODRÍGUEZ-MARTÍNEZ AB, ARCE A, CARRERA N ET AL. Phenotypic variability in familial prion diseases due to the D178N mutation. J Neurol Neurosurg Psychiatry 2005; 76: 1491-1496.
85. LEE HS, GOLDFARB LG. Global distribution of fatal familial insomnia: founder or recurrent mutations. Neurogenetics 2008; 9: 301-302.
86. RODRÍGUEZ-MARTÍNEZ AB, BARREAU C, COUPRY I, YAGUE J, SÁNCHEZ-VALLE R, GALDOS-ALCELAY L ET AL. Ancestral origins of the prion protein gene D178N mutation in the Basque Country. Hum Genet 2005; 117: 61-69.
87. AYUSO T, TUNON T, ERRO ME. Sleep disorders in prion diseases. An Sist Sanit Navar 2007; 30 Suppl 1: 135-141.
88. MONTAGNA P, GAMBETTI P, CORTELLI P, LUGARESI E. Familial and sporadic fatal insomnia. Lancet Neurol 2003; 2: 167-176.
89. KNIGHT R. The diagnosis of prion diseases. Parasitology 1998; 117 (Suppl.): S3-11.
90. BAXTER HC, FRASER JR, LIU WG, FORSTER JL, CLOKIE S, STEINACKER P ET AL. Specific 14-3-3 isoform detection and immunolocalization in prion diseases. Biochem Soc Trans 2002; 30: 387-391.
91. GMITTEROVA K, HEINEMANN U, BODEMER M, KRASNIANSKI A, MEISSNER B, KRETZSCHMAR HA ET AL. 14-3-3 CSF levels in sporadic Creutzfeldt-Jakob disease differ across molecular subtypes. Neurobiol Aging 2009; 30: 1842-1850.
92. LADOGANA A, SÁNCHEZ-JUAN P, MITROVA E, GREEN A, CUADRADO-CORRALES N, SÁNCHEZ-VALLE R ET AL. Cerebrospinal fluid biomarkers in human genetic transmissible spongiform encephalopathies. J Neurol 2009; 256: 1620-1628.
93. POSER S, MOLLENHAUER B, KRAUBETA A, ZERR I, STEINHOFF BJ, SCHROETER A ET AL. How to improve the clinical diagnosis of Creutzfeldt-Jakob disease. Brain 1999; 122: 2345-2351.
94. WILKER EW, GRANT RA, ARTIM SC, YAFFE MB. A structural basis for 14-3-3sigma functional specificity. J Biol Chem 2005; 280: 18891-18898.
95. ZEIDLER M. 14-3-3 cerebrospinal fluid protein and Creutzfeldt-Jakob disease. Ann Neurol 2000; 47: 683-684.
96. BOESENBERG-GROSSE C, SCHULZ-SCHAEFFER WJ, BODEMER M, CIESIELCZYK B, MEISSNER B, KRASNIANSKI A ET AL. Brain-derived proteins in the CSF: do they correlate with brain pathology in CJD? BMC Neurol 2006: 6-35.
97. XIANG W, WINDL O, WUNSCH G, DUGAS M, KOHLMANN A, DIERKES N ET AL. Identification of differentially expressed genes in scrapie-infected mouse brains by using global gene expression technology. J Virol 2004; 78: 11051-11060.
98. SÁNCHEZ-JUAN P, BISHOP MT, GREEN A, GIANNATTASIO C, ARIAS-VÁSQUEZ A, POLEGGI A ET AL. No evidence for association between tau gene haplotypic variants and susceptibility to Creutzfeldt-Jakob disease. BMC Med Genet 2007: 8-77.
99. OTTO M, WILTFANG J, CEPEK L, NEUMANN M, MOLLENHAUER B, STEINACKER P ET AL. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2002; 58: 192-197.
100. KROPP S, ZERR I, SCHULZ-SCHAEFFER WJ, RIEDEMANN C, BODEMER M, LASKE C ET AL. Increase of neuron-specific enolase in patients with Creutzfeldt-Jakob disease. Neurosci Lett 1999; 26: 124-126.
101. ZERR I, BODEMER M, OTTO M, POSER S, WINDL O, KRETZSCHMAR HA ET AL. Diagnosis of Creutzfeldt-Jakob disease by two-dimensional gel electrophoresis of cerebrospinal fluid. Lancet 1996; 348: 846-849.
102. BLUMENKRON D, GUERRERO P, RAMIRO M. Creutzfeldt-Jakob Disease. Medicina Interna Mexicana 2007; 23: 34-46.
103. WIESER HG, SCHINDLER K, ZUMSTEG D. EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol 2006; 117: 935-951.
104. ZEIDLER M, WILL RG, IRONSIDE JW, SELLAR R, WARDLAW J. Creutzfeldt-Jakob disease and bovine spongiform encephalopathy. Magnetic resonance imaging is not a sensitive test for Creutzfeldt-Jakob disease. BMJ 1996; 312: 844.
105. COLLINS SJ, SÁNCHEZ-JUAN P, MASTERS CL, KLUG GM, VAN DUIJN C, POLEGGI A ET AL. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006; 129: 2278-2287.
106. LODI R, PARCHI P, TONON C, MANNERS D, CAPELLARI S, STRAMMIELLO R ET AL. Magnetic resonance diagnostic markers in clinically sporadic prion disease: a combined brain magnetic resonance imaging and spectroscopy study. Brain 2009; 132: 2669-2679.
107. IWASAKI Y, IGARASHI O, ICHIKAWA Y, IKEDA K. Sporadic Creutzfeldt-Jakob disease: magnetic resonance imaging and clinical findings. Neurology 2005; 64: 1318.
108. MORENO-IZCO F, MARTÍNEZ-GIL A. Creutzfeldt-Jakob disease: alterations in an isolated cortical signal in diffusion magnetic resonance imaging. Rev Neurol 2005; 40: 38-42.
109. MACFARLANE RG, WROE SJ, COLLINGE J, YOUSRY TA, JAGER HR. Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry 2007; 78: 664-670.
110. MORENO-IZCO F, ARRIOLA-LARRARTE L. Neuroimaging in the diagnosis of human transmissible spongiform encephalopathies. Neurología 2006; 21: 428-436.
111. CASTILLO M, THURNHER M. Imaging viral and prion infections. Semin Roentgenol 2004; 39: 482-494.
112. TABER KH, CORTELLI P, STAFFEN W, HURLEY RA. The expanding role of imaging in prion disease. J Neuropsychiatry Clin Neurosci 2002; 14: 371-376.
113. MEAD S. Prion disease genetics. Eur J Hum Genet 2006; 14: 273-281.
114. IRONSIDE JW, RITCHIE DL, HEAD MW. Phenotypic variability in human prion diseases. Neuropathol Appl Neurobiol 2005; 31: 565-579.
115. KOBAYASHI A, HIZUME M, TERUYA K, MOHRI S, KITAMOTO T. Heterozygous inhibition in prion infection: the stone fence model. Prion 2009; 3: 27-30.
116. TREVITT CR, COLLINGE J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129: 2241-2265.
117. MAY BC, FAFARMAN AT, HONG SB, ROGERS M, DEADY LW, PRUSINER SB ET AL. Potent inhibition of scrapie prion replication in cultured cells by bis-acridines. Proc Natl Acad Sci USA 2003; 100: 3416-3421.
118. DOH-URA K, IWAKI T, CAUGHEY B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000; 74: 4894-4897.
119. FAROOQUI AA, ONG WY, HORROCKS LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev 2006; 58: 591-620.
120. TSUBOI Y, DOH-URA K, YAMADA T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology 2009; 29: 632-636.
121. PARRY A, BAKER I, STACEY R, WIMALARATNA S. Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulphate. J Neurol Neurosurg Psychiatry 2007; 78: 733-734.
122. DOH-URA K, ISHIKAWA K, MURAKAMI-KUBO I, SASAKI K, MOHRI S, RACE R ET AL. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol 2004; 78: 4999-5006.
123. CAUGHEY B, RAYMOND GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol 1993; 67: 643-650.
124. WEBB S, LEKISHVILI T, LOESCHNER C, SELLARAJAH S, PRELLI F, WISNIEWSKI T ET AL. Mechanistic insights into the cure of prion disease by novel antiprion compounds. J Virol 2007; 81: 10729-10741.
125. FENG BY, TOYAMA BH, WILLE H, COLBY DW, COLLINS SR, MAY BC ET AL. Small-molecule aggregates inhibit amyloid polymerization. Nat Chem Biol 2008; 4: 197-199.
126. RUDYK H, VASILJEVIC S, HENNION RM, BIRKETT CR, HOPE J, GILBERT IH. Screening Congo Red and its analogues for their ability to prevent the formation of PrP-res in scrapie-infected cells. J Gen Virol 2000; 81: 1155-1164.
127. CAUGHEY B, ERNST D, RACE RE. Congo red inhibition of scrapie agent replication. J Virol 1993; 67: 6270-6272.
128. INGROSSO L, LADOGANA A, POCCHIARI M. Congo red prolongs the incubation period in scrapie-infected hamsters. J Virol 1995; 69: 506-508.
129. POCCHIARI M, SCHMITTINGER S, MASULLO C. Amphotericin B delays the incubation period of scrapie in intracerebrally inoculated hamsters. J Gen Virol 1987; 68: 219-223.
130. MANGE A, NISHIDA N, MILHAVET O, MCMAHON HE, CASANOVA D, LEHMANN S. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J Virol 2000; 74: 3135-3140.
131. ADJOU KT, DEMAIMAY R, LASMEZAS C, DESLYS JP, SEMAN M, DORMONT D. MS-8209, a new amphotericin B derivative, provides enhanced efficacy in delaying hamster scrapie. Antimicrob Agents Chemother 1995; 39: 2810-2812.
132. ADJOU KT, DEMAIMAY R, DESLYS JP, LASMEZAS CI, BERINGUE V, DEMART S ET AL. MS-8209, a water-soluble amphotericin B derivative, affects both scrapie agent replication and PrPres accumulation in Syrian hamster scrapie. J Gen Virol 1999; 80: 1079-1085.
133. DEMAIMAY R, ADJOU KT, BERINGUE V, DEMART S, LASMEZAS CI, DESLYS JP ET AL. Late treatment with polyene antibiotics can prolong the survival time of scrapie-infected animals. J Virol 1997; 71: 9685-9689.
134. MCKENZIE D, KACZKOWSKI J, MARSH R, AIKEN J. Amphotericin B delays both scrapie agent replication and PrP-res accumulation early in infection. J Virol 1994; 68: 7534-7536.
135. SOLER L, CAFFREY P, MCMAHON HE. Effects of new amphotericin analogues on the scrapie isoform of the prion protein. BIOCHIM BIOPHYS ACTA 2008; 1780: 1162-1167.
136. ADJOU KT, DESLYS JP, DEMAIMAY R, DORMONT D. Probing the dynamics of prion diseases with amphotericin B. Trends Microbiol 1997; 5: 27-31.
137. COLLINGE J, GORHAM M, HUDSON F, KENNEDY A, KEOGH G, PAL S ET AL. Safety and efficacy of quinacrine in human prion disease (Prion-1 study): a patient-preference trial. Lancet Neurol 2009; 8: 334-344.
138. GAYRARD V, PICARD-HAGEN N, VIGUIE C, LAROUTE V, ANDREOLETTI O, TOUTAIN PL. A possible pharmacological explanation for quinacrine failure to treat prion diseases: pharmacokinetic investigations in a ovine model of scrapie. Br J Pharmacol 2005; 144: 386-393.
139. DOHGU S, YAMAUCHI A, TAKATA F, SAWADA Y, HIGUCHI S, NAITO M ET AL. Uptake and efflux of quinacrine, a candidate for the treatment of prion diseases, at the blood-brain barrier. Cell Mol Neurobiol 2004; 24: 205-217.
140. GHAEMMAGHAMI S, AHN M, LESSARD P, GILES K, LEGNAME G, DEARMOND SJ ET AL. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog 2009; 5: e1000673.
141. BARRET A, TAGLIAVINI F, FORLONI G, BATE C, SALMONA M, COLOMBO L ET AL. Evaluation of quinacrine treatment for prion diseases. J Virol 2003; 77: 8462-8469.
142. KORTH C, MAY BC, COHEN FE, PRUSINER SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA 2001; 98: 9836-9841.
143. TAGLIAVINI F, FORLONI G, COLOMBO L, ROSSI G, GIROLA L, CANCIANI B ET AL. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J Mol Biol 2000; 300: 1309-1322.
144. FORLONI G, IUSSICH S, AWAN T, COLOMBO L, ANGERETTI N, GIROLA L ET AL. Tetracyclines affect prion infectivity. Proc Natl Acad Sci USA 2002; 99: 10849-10854.
145. DE LUIGI A, COLOMBO L, DIOMEDE L, CAPOBIANCO R, MANGIERI M, MICCOLO C ET AL. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS One 2008; 3: e1888.
146. LOVE R. Old drugs to treat new variant Creutzfeldt-Jakob disease. Lancet 2001; 358: 563.
147. MARELLA M, LEHMANN S, GRASSI J, CHABRY J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J Biol Chem 2002; 277: 25457-25464.
148. ARRUDA WO, BORDIGNON KC, MILANO JB, RAMINA R. Creutzfeldt-Jakob disease, Heidenhain variant: case report with MRI (DWI) findings. Arq Neuropsiquiatr 2004; 62: 347-352.
149. DEES C, WADE WF, GERMAN TL, MARSH RF. Inactivation of the scrapie agent by ultraviolet irradiation in the presence of chlorpromazine. J Gen Virol 1985; 66: 845-849.
150. CRONIER S, BERINGUE V, BELLON A, PEYRIN JM, LAUDE H. Prion strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J Virol 2007; 81: 13794-13800.
151. KORTH C, PETERS PJ. Emerging pharmacotherapies for Creutzfeldt-Jakob disease. Arch Neurol 2006; 63: 497-501.
152. PERRIER V, SOLASSOL J, CROZET C, FROBERT Y, MOURTON-GILLES C, GRASSI J ET AL. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem 2004; 89: 454-463.
153. FOLLETTE P. New perspectives for prion therapeutics meeting. Prion disease treatment's early promise unravels. Science 2003; 299: 191-192.
154. PARCHI P, STRAMMIELLO R, NOTARI S, GIESE A, LANGEVELD JP, LADOGANA A ET AL. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol 2009; 118: 659-671.
155. PETCHANIKOW C, SABORIO GP, ANDERES L, FROSSARD MJ, OLMEDO MI, SOTO C. Biochemical and structural studies of the prion protein polymorphism. FEBS Lett 2001; 509: 451-456.
156. TAHIRI-ALAOUI A, SIM VL, CAUGHEY B, JAMES W. Molecular heterosis of prion protein beta-oligomers. A potential mechanism of human resistance to disease. J Biol Chem 2006; 281: 34171-34178.
157. LAWSON VA, COLLINS SJ, MASTERS CL, HILL AF. Prion protein glycosylation. J Neurochem 2005; 93: 793-801.
158. Vorberg I, Priola SA. Molecular basis of scrapie strain glycoform variation. J Biol Chem 2002; 277: 36775-36781.
159. HILL AF, JOINER S, BECK JA, CAMPBELL TA, DICKINSON A, POULTER M ET AL. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain 2006; 129: 676-685.
160. KOVACS GG, TRABATTONI G, HAINFELLNER JA, IRONSIDE JW, KNIGHT RS, BUDKA H. Mutations of the prion protein gene phenotypic spectrum. J Neurol 2002; 249: 1567-1582.
161. HARRIS DA, TRUE HL. New insights into prion structure and toxicity. Neuron 2006; 50: 353-357.
162. HODAK M, CHISNELL R, LU W, BERNHOLC J. Functional implications of multistage copper binding to the prion protein. Proc Natl Acad Sci USA 2009; 106: 11576-11581.
163. BROWN DR. Prion protein expression modulates neuronal copper content. J Neurochem 2003; 87: 377-385.
164. KANAANI J, PRUSINER SB, DIACOVO J, BAEKKESKOV S, LEGNAME G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem 2005; 95: 1373-1386.
165. HOUNSELL EF. Prions in control of cell glycosylation. Biochem J 2004; 380: e5-e6.
166. LINDEN R, MARTINS VR, PRADO MA, CAMMAROTA M, IZQUIERDO I, BRENTANI RR. Physiology of the prion protein. Physiol Rev 2008; 88: 673-728.
167. ZOMOSA-SIGNORET V, ARNAUD JD, FONTES P, ÁLVAREZ-MARTÍNEZ MT, LIAUTARD JP. Physiological role of the cellular prion protein. Vet Res 2008; 39: 9.
168. WATTS JC, WESTAWAY D. The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta 2007; 1772: 654-672.